Rev Med UAS
Rev Med UAS; Vol. 15 No. 3. Julio-Septiembre 2025
ISSN 2007-8013
Darihán María Sánchez-Castro1*, Keren Adasa Ruiz-de la Cruz2, Raúl Alfonso Ruiz-Barraza3, Efraín Romo-García4, Alonzo Meza-Anguiano5
*Autor de correspondencia: Darihán María Sánchez Castro
Domicilio: Calle Eustaquio Buelna No. 91, Col. Gabriel Leyva, C.P 80030 Culiacán, Sinaloa.
Correo: Darihansc@gmail.com
DOI http://dx.doi.org/10.28960/revmeduas.2007-8013.v15.n3.007
Texto Completo PDFRecibido 1 de marzo 2025, aceptado 3 de junio 2025
RESUMEN
Antecedentes: El hamartoma astrocítico es una neoplasia glial benigna, comúnmente encontrada en pacientes pediátricos o adolescentes. En más de la mitad de los casos se asocia a síndromes sistémicos hereditarios, siendo la esclerosis tuberosa el más común. Su impacto visual depende de la localización, y aunque muchas lesiones son asintomáticas, algunas pueden desarrollar importantes complicaciones. --- Caso clínico: Paciente femenino de 10 años de edad traído a consulta por dificultad visual en el ámbito escolar. A la exploración oftalmológica se identificó una lesión multilobulada en la retina periférica del ojo derecho, de aspecto amarillento y sobreelevado. Los estudios de imagen multimodal confirmaron hallazgos compatibles con un hamartoma astrocítico tipo III. Se realizó valoración por los servicios de neurología, nefrología y psicología, quienes descartaron asociación a complejo de esclerosis tuberosa u otras facomatosis sistémicas. --- Conclusiones: El hallazgo de un hamartoma astrocítico retiniano debe motivar la búsqueda intencionada de enfermedades sistémicas. Los estudios de imagen multimodales son esenciales para el diagnóstico y caracterización de la neoplasia. Este caso enfatiza la importancia de un abordaje multidisciplinario y seguimiento estrecho en pacientes con neoplasias retinianas atípicas.
Palabras clave: hamartoma astrocítico, facomatosis, esclerosis tuberosa, oftalmología pediátrica.
ABSTRACT
Background: Astrocytic hamartoma is a benign glial neoplasm commonly found in pediatric or adolescent patients. In more than half of the cases, it is associated with hereditary systemic syndromes, the most common being tuberous sclerosis. Its visual impact depends on the location, and although many lesions are asymptomatic, some can develop significant complications. --- Clinical case: A 10-year-old female was brought to the clinic for visual impairment at school. Ophthalmologic examination identified a multilobulated lesion in the peripheral retina of the right eye, yellowish in appearance and elevated. Multimodal imaging studies confirmed findings compatible with a type III astrocytic hamartoma. A multidisciplinary evaluation ruled out association with tuberous sclerosis complex or other systemic phakomatoses. --- Conclusion: The discovery of a retinal astrocytic hamartoma should prompt a comprehensive search for systemic diseases. Multimodal imaging studies are essential for the diagnosis and characterization of the neoplasm. This case emphasizes the importance of an interdisciplinary approach and close follow-up in patients with atypical retinal neoplasms.
Keywords: astrocytic hamartoma, phakomatoses, tuberous sclerosis, pediatric ophthalmology.
Introducción
El hamartoma astrocítico retiniano (HAR) es una neoplasia glial benigna que se origina a partir de los astrocitos de la retina. Es más frecuente en pacientes pediátricos o adolescentes, y se encuentra hasta en un 50% de los pacientes con complejo de esclerosis tuberosa (CET), constituyendo el hallazgo oftalmológico más común. El CET es una facomatosis hereditaria rara, caracterizada por manifestaciones sistémicas variables en ojos, cerebro, riñones, pulmones y piel1. En este contexto, los HAR pueden presentarse de forma bilateral y multifocal en hasta el 50% de los casos, mientras que en su forma esporádica, que representa aproximadamente el 29%, suelen ser lesiones solitarias2. Clínicamente, el HAR se manifiesta como una masa amarillenta-grisácea, con vasos retinianos mínimamente dilatados, tracción retiniana fina y calcificaciones intrínsecas. Su impacto visual depende de la localización, siendo más frecuentes las lesiones yuxtapapilares o en retina periférica3. El diagnóstico es principalmente clínico, aunque las herramientas de imagen, como la tomografía de coherencia óptica (OCT), han permitido una mejor caracterización. Según su morfología en OCT, se describen cuatro tipos: tipo I (lesión plana), tipo II (elevación leve con tracción), tipo III (áreas “apolilladas” con calcificación) y tipo IV (cavidades intralesionales vacías)4. Aunque muchas lesiones son asintomáticas, especialmente si no comprometen la mácula, pueden desarrollarse importantes complicaciones como hemorragia vítrea, siembra vítrea, o alteraciones vasculares retinianas5. El reconocimiento oportuno de esta entidad es importante, ya que obliga a realizar un abordaje integral que descarte su asociación con otros síndromes sistémicos.
Presentación de caso
Se presenta paciente femenino de 10 años de edad, traída a consulta por presentar dificultades visuales en la escuela. Al interrogatorio dirigido, la madre refiere retraso leve en el neurodesarrollo, sin antecedentes de convulsiones, afecciones renales, cardiacas, ni cutáneas. Respecto a los antecedentes heredofamiliares, refiere que el padre de la paciente falleció en un accidente automovilístico, y que presentaba crisis epilépticas sin diagnóstico establecido. Durante la valoración inicial se diagnosticó astigmatismo miópico compuesto en ambos ojos, indicándose corrección óptica con lentes. La agudeza visual mejor corregida fue de 20/20 en ambos ojos. Refracción bajo cicloplejia: ojo derecho (OD) -3.00 -1.00 x 90 y ojo izquierdo (OI) -2.50 -0.50 x 60. A la exploración oftalmológica bajo lámpara de hendidura, se observó segmento anterior sin alteraciones en ambos ojos. En el fondo de ojo del OD se identificó una lesión única en retina periférica inferonasal.
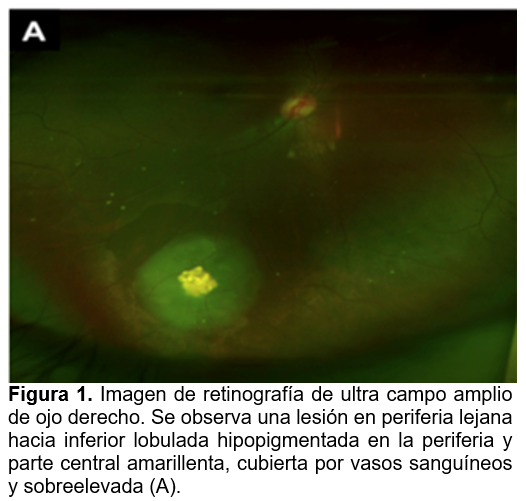
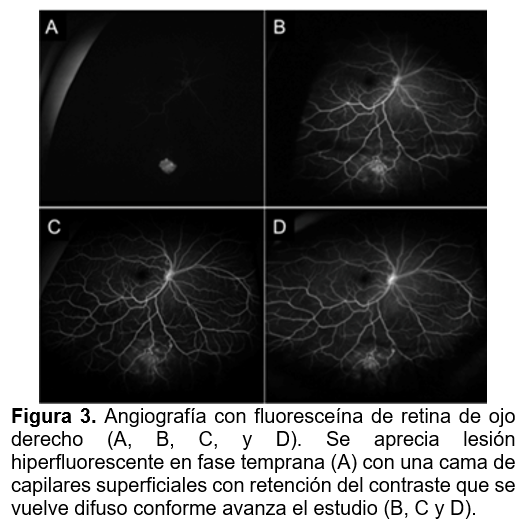
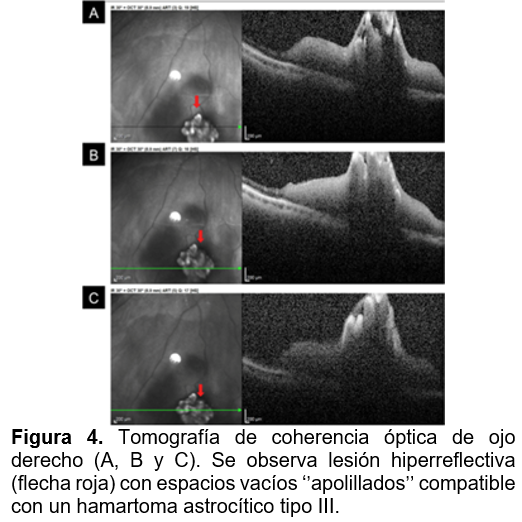
Se realizaron estudios de imagen multimodal con cámara de fondo de ojo Optos. En el OD, la retinografía reveló en el cuadrante inferonasal una lesión multilobulada, hipopigmentada, con aspecto de moras, cubierta por vasos sanguíneos, sobreelevada, y acompañada de hialosis asteroidea (Figura 1). En autofluorescencia se observa hiper autofluorescencia franca de la lesión, con diferenciación clara de los lóbulos calcificados (figura 2). La angiografía con fluoresceína mostró hiperfluorescencia temprana con una cama capilar superficial, y retención progresiva del contraste, que se volvió difusa en fases tardías (figura 3). En el estudio con tomografía de coherencia óptica (OCT), se evidenció una lesión hiperreflectiva con espacios ópticamente vacíos tipo “apolillados”, compatibles con hamartoma astrocítico tipo III. Se observó transición de la retina normal a un engrosamiento confinado a la retina interna, así como edema leve circundante a la lesión en domo (figura 4). El ojo izquierdo no mostró alteraciones en ningún estudio. Se realizó interconsulta a los servicios de psicología, neurología y nefrología para completar la valoración sistémica y descartar asociación con complejo de esclerosis tuberosa.

Discusión
El hamartoma astrocítico retiniano (HAR) es una neoplasia benigna de origen glial que puede presentarse de forma hereditaria o adquirida. Su variante hereditaria se asocia con mayor frecuencia al complejo de esclerosis tuberosa (CET), un trastorno genético causado por mutaciones en los genes supresores de tumor TSC1 y TSC2, que codifican las proteínas hamartina y tuberina, respectivamente. El CET, anteriormente conocido como facomatosis de Pringle-Bourneville, presenta una incidencia estimada de 1 en cada 6,000 a 10,000 nacidos vivos6. Entre sus manifestaciones sistémicas destacan alteraciones neurológicas como epilepsia, espasmos infantiles, retraso psicomotor y trastornos del espectro autista, así como lesiones cutáneas, renales, pulmonares y cardíacas. Por otro lado, los HAR solitarios o adquiridos representan aproximadamente el 29 % de los casos y suelen reportarse con menor frecuencia en la literatura2. Estas lesiones generalmente son asintomáticas, aunque pueden afectar el campo visual dependiendo de su localización, siendo más comunes las lesiones adyacentes al nervio óptico y en la retina periférica. Clínicamente se presentan como lesiones únicas, multilobuladas, de aspecto esponjoso o en forma de mora, con calcificaciones intralesionales observadas en hasta el 50 % de los casos7. Pueden presentar complicaciones como hemorragias vítreas y exudado subretiniano, manifestaciones que son más frecuentes en comparación con los HAR hereditarios5. El diagnóstico diferencial del HAR incluye lesiones como melanoma coroideo amelanótico, metástasis de melanoma, retinoblastoma y osteoma coroideo8. No obstante, el desarrollo de técnicas de imagen multimodal, especialmente OCT y la angiografía con fluoresceína, han permitido una mejor caracterización estructural y vascular de estas lesiones, facilitando su diferenciación clínica y diagnóstico oportuno.
Conclusión
El hamartoma astrocítico retiniano es una entidad benigna y poco frecuente, cuyo hallazgo, especialmente en pacientes sin diagnóstico previo de enfermedades sistémicas, debe motivar una valoración multidisciplinaria para descartar su asociación con esclerosis tuberosa, entre otras facomatosis. Las herramientas de imagen multimodal, en particular la tomografía de coherencia óptica y la angiografía con fluoresceína, son fundamentales para su caracterización y diagnóstico diferencial. En el caso presentado, no se identificaron hallazgos compatibles con esclerosis tuberosa ni con otras patologías sistémicas, por lo que se estableció el diagnóstico de hamartoma astrocítico solitario. Se indicó seguimiento oftalmológico semestral para vigilar la estabilidad de la lesión. Este caso enfatiza la importancia de una exploración oftalmológica integral en pacientes pediátricos con síntomas visuales inespecíficos, así como la necesidad de un abordaje multidisciplinario para la detección oportuna de enfermedades sistémicas subyacentes.
Referencias