
Rev Med UAS
Vol. 13: No. 1. Enero-Marzo 2023
ISSN 2007-8013
Joshua Pérez-Velarde1, Nallely Duarte-Iribe2, Anais Álvarez-Villarreal3.
* Correspondencia: Joshua Pérez-Velarde
Calle Heroico colegio militar, col. 5 de mayo. Culiacán Sinaloa, México.
Correo; joshuapv20@gmail.com
DOI http://dx.doi.org/10.28960/revmeduas.2007-8013.v13.n1.008
Texto Completo PDFRecibido 16 de febrero de 2021, aceptado 01 de febrero de 2022
RESUMEN
La enfermedad de Fabry (EF) es un trastorno del metabolismo de los glucoesfingolípidos, ligado al cromosoma X, recesivo y poco frecuente, que resulta de una deficiencia en la hidrolasa alfa-galactosidasa A (α-GalA) causada por una variante del gen GLA de afección multisistémica y que produce disfunción orgánica. La afectación ocular más típica de la EF es la córnea verticillata. Reportamos el caso de una paciente de 34 años de edad diagnosticada con EF de novo. Se realizó evaluación oftalmológica previa al inicio de su terapia de reemplazo enzimática (TRE). Como hallazgo se encontró córnea verticillata en ojo izquierdo y tortuosidad de vasos conjuntivales, los cuales mostraron regresión tres meses posteriores a su TRE con alfa galactosidasa.
Palabras clave: Enfermedad de Fabry, Córnea verticillata, Terapia remplazo enzimático.
ABSTRACT
Fabry disease (FD) is a rare, X-linked, recessive disorder of glycosphingolipid metabolism, resulting from a deficiency in alpha-galactosidase A (α-GalA) hydrolase caused to a variant of the GLA gene, ocurring as a multisystemic affection and producing organ dysfunction. The most typical ocular involvement of FD is the cornea verticillate. We report the case of a 34-year-old patient recently diagnosed with FD. An ophthalmological evaluation was performed prior to the start of enzyme replacement therapy (ERT). As a finding, cornea verticillate was found in the left eye and tortuosity of the conjunctival in both eyes, both of them showed regression three months after ERT with alpha galactosidase.
Keywords: Fabry disease, Cornea verticillata, Enzyme replacement therapy.
INTRODUCCIÓN
La enfermedad de Fabry (EF) es una enfermedad poco común ligada al cromosoma X. Ocasionada por la deficiencia parcial o completa de la enzima lisosomal alfa-galactosidasa A (a-Gal A) responsable del catabolismo de los esfingolípidos. Es altamente penetrante en los hombres y de variabilidad expresiva en las mujeres. La prevalencia oscila entre 1:40.000 y 1:117.000 individuos1. La EF presenta daño multiorgánico, como manifestaciones cardiacas, enfermedad renal, acroparestesias, angioqueratomas, y a nivel oftalmológico el signo más frecuente y común es la córnea verticillata2-3. El tratamiento es la terapia de reemplazo enzimático (ERT), ya sea Replagal ® (Agalsidasa alfa) o Fabrazyme ® (Agalsidasa beta), y desde hace unos años la terapia con chaperonas Migalastat4.
En el presente caso describimos los hallazgos oftalmológicos de una paciente con carga genética heterocigota y las diferencias que hubo antes y después de haber recibido la terapia con reemplazo enzimático.
PRESENTACIÓN DE CASO
Se evaluó a una paciente femenina de 34 años de edad. Con antecedentes familiares de hipertensión arterial, diabetes mellitus tipo 2 y complicaciones renales y cardiológicas en línea familiar secundario a crónico degenerativas.
Refirió antecedente de enfermedad inflamatoria intestinal de 15 años de evolución sin tratamiento, dificultad para concebir embarazo, miopía diagnosticada a los 15 años de edad, taquicardias esporádicas al momento de acostarse y enfermedad de Fabry diagnosticada en 2019. Su tratamiento farmacológico fue a base de telmisartán, atorvastatina, enalapril y agalsidasa beta. Menciona sintomatología durante su infancia con intolerancia al calor y ardor en sus manos. En 2019 acude a valoración por nefrología presentando edema de miembros inferiores y proteinuria de 2,500 mg. Se le realizó biopsia renal donde se reportó esclerosis focal y segmentaria, nefritis tubulointersticial crónica y ateroesclerosis avanzada con fibrosis intersticial en bandas. Debido a la alta sospecha de afección renal por EF se realizó estudio genético en agosto de 2019 donde se obtuvo el resultado diagnóstico de variante patogénica en heterocigosis en el gen GLA c.898C, con concentración incrementada de LISO-GB3 de 7.8 ng/ml (valor de referencia < 1.8 ng/ml). La paciente inició protocolo de estudio, a nivel dermatológico se identificaron lesiones hipodensas blancas y rosas compatibles con angioqueratomas de 1 año de evolución. Neurológica y cardiovascularmente no se encontraron alteraciones.
Valorada por oftalmología en junio de 2021, se le realizó exploración en lámpara de hendidura y fotografías de fondo de ojo. En ojo derecho tuvo una capacidad visual (CV) de 20/40, ojo izquierdo con CV de 20/25. Presiones intraoculares de 14 mmHg en ambos ojos observando conjuntiva de ojo derecho con pinguécula nasal y temporal y vasos tortuosos. La córnea se encontraba clara, y con presencia de queratitis punteada superficial (QPS) en su tercio inferior. La cámara anterior estaba profunda y bien formada, van Herick de IV, iris café homogéneo y cristalino sin signos de opacidad El fondo de ojo se observó con vítreo claro, retina aplicada y sin alteraciones a nivel macular, una papila óptica de adecuado color, un buen contorno, y relación papila/excavación en su diámetro vertical de 30%, los vasos se encontraban tortuosos en las arcadas temporal y nasal en todo el polo posterior. En ojo izquierdo, se documenta una pinguécula en conjuntiva nasal y temporal con vasos tortuosos conjuntivales. En la córnea se encontraba depósitos en epitelio de conformación arremolinada (Figura 1) y a la tinción con fluoresceína QPS de predominio en tercio inferior. Sin opacidades en cristalino y resto de segmento anterior normal. El fondo de ojo presentó vítreo claro, retina aplicada, mácula sin alteraciones, papila óptica sin anormalidades, con vasos tortuosos en las arcadas temporal y nasal y un cruce arteriovenoso en la arteria temporal inferior. En esa consulta se integró el diagnóstico de síndrome de ojo seco y se le indicó el uso de lubricantes sin conservador.
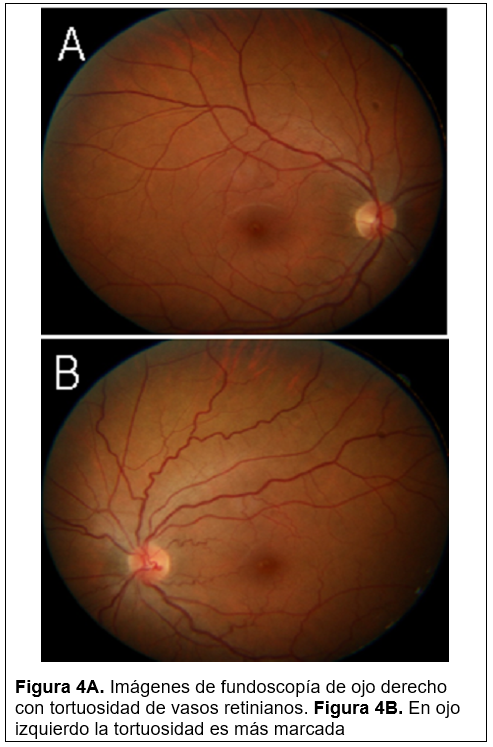
En marzo de 2022 acude a cita subsecuente, se encontraba en su octavo ciclo de Agalsidasa beta con una dosis de 65 mg intravenoso, el cual se le administraba cada 14 días, en dicha revisión mantenía la misma CV en ambos ojos. En segmento anterior de ambos ojos se encontraba tortuosidad de vasos conjuntivales de mayor predominio en ojo izquierdo (Figura 2A y f’igura 2B), En la exploración de córnea en ojo derecho se encontraba clara y sin QPS, sin depósitos en epitelio y en ojo izquierdo se encontraba clara, sin QPS y con depósitos epiteliales casi imperceptibles (Figura 3). A nivel de fondo de ojo la exploración fue similar a su consulta anterior, manteniéndose los vasos tortuosos en ambos ojos, con predominio en el ojo izquierdo (Figura 4A y figura 4B)


DISCUSIÓN
La deficiencia de la enzima lisosomal alfa-galactosidasa (a-GAL) da como resultado la acumulación progresiva de globotriaosilceramida (Gb-3) en tejido vascular, con diversos grados de afectación de forma, sistémica. El efecto de Gb-3 a nivel ocular produce córnea verticillata, siendo el signo más característico y común de Fabry a nivel ocular5. Se han identificado 3 tipos de fenotipos: Una variedad clásica, casi reservada a masculinos sin actividad de a-GAL, una variedad caracterizada por masculinos con manifestaciones atípicas, de inicio en la infancia, y una presentación en pacientes femeninas, con manifestaciones variables, esto por la penetrancia genética variable de la paciente6. En general, se acepta que las pacientes femeninas son heterocigotos y pueden heredar la mutación de cualquiera de sus padres6
Debido a su componente genético las manifestaciones clínicas pueden variar con la edad, referido por nuestra paciente como una sensación de quemazón o ardor en manos y pies (acroparestesia), el caso cursó con dolor neuropático crónico, el cual en la tercera década de la vida puede progresar a fenómenos con disautonomía7. La afección renal puede tener un inicio temprano8, reportándose en nuestra paciente daño renal avanzado con esclerosis glomerular focal y fibrosis intersticial, manifestación inicial documentada en la paciente, secundaria al edema de miembros inferiores y proteinuria. A nivel dermatológico existe daño debido a las paredes capilares debilitadas se pueden presentar ectasias vasculares conocidas como angioqueratomas9
La córnea verticillata es producida por los depósitos de GB-3 a nivel de la membrana basal epitelial y la membrana de Bowman, haciendo ese patrón “en vórtice” de color blanco o dorado. Se ha planteado que la presencia de afección corneal está relacionada con una mayor severidad del padecimiento2, aunque usualmente no afecta la agudeza visual. En cristalino el depósito de GB-3 también puede opacificar a nivel subcapsular posterior y en las suturas corticales con disposición radial, produciendo una catarata de Fabry10, nuestro caso solo presentó depósitos visibles de GB-3 compatibles con cornea verticillata. Los vasos conjuntivales además, se vieron afectados en nuestro caso, descrito en la literatura por una acumulación de Gb-3 en el endotelio vascular, presentándose como tortuosidades a nivel conjuntival y retiniano11.
El tratamiento específico seleccionado en nuestra paciente para la EF consistió en terapia de reemplazo enzimático con agalsidasa beta4. Evaluamos sus características oftálmicas después del inicio de la terapia de reemplazo enzimática, que fue durante el octavo ciclo de su tratamiento con 65 mg IV de agalsidasa beta cada 2 semanas y el uso de inhibidores de la enzima convertidora de angiotensina para limitar su proteinuria. Encontramos que los depósitos corneales en el ojo izquierdo se habían resuelto notablemente, mientras que en la vasculatura conjuntival existía una mejoría parcial, sin otro cambio oftalmológico evidente.
En las investigaciones médicas relacionadas a biomarcadores para seguimiento y monitorización de la EF se han realizado estudios con diversas estrategias. A nivel sérico, para el seguimiento de los pacientes se utiliza la determinación sérica de lyso-GB3 13-14. A nivel oftalmológico el seguimiento de los pacientes ha sido con la tomografía de coherencia óptica (TCO) y sus diversas modalidades, con enfoque en el grosor retiniano y la cuantificación de la variabilidad de la irrigación retiniana pero ninguna publicación ha analizado a los pacientes posterior a su inicio con TRE15-17. En cuanto a la córnea no se ha realizado investigación relacionada a la mejoría de la córnea verticillata posterior a la TRE.
En este caso clínico se documenta a una paciente con una presentación heterocigótica de la enfermedad de novo, en la cual se observó en un inicio los hallazgos oculares más comunes reportados en la literatura médica y la regresión que presentaron posterior al inicio de la terapia de sustitución enzimática, tales como la mejoría de la distribución de la córnea verticillata en ojo izquierdo y la normalización de la vasculatura conjuntival.
No contamos con literatura científica mexicana de otro caso similar, por lo cual consideramos importante compartirlo. Asimismo, este reporte clínico destaca el hecho de que un hallazgo tal como una córnea verticillata puede ser un punto clave para sospechar de una Enfermedad de Fabry18 y un probable biomarcador para su seguimiento.
Responsabilidades éticas Protección de personas y animales
Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos
Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informado
Los autores declaran que en este artículo no aparecen datos de pacientes.
Referencias