Rev Med UAS
Vol. 12: No. 4. Octubre-Diciembre 2022
ISSN 2007-8013
Juan Rosales-Martínez1, Manlio F. Lara-Duck2, Antonio Gutiérrez-Sierra3, Netzahualcóyotl Mayek-Pérez3*
* Correspondencia: Netzahualcóyotl Mayek-Pérez
Primera S/N, col. El Círculo. Reynosa Tamaulipas, México.
Correo electrónico: nmayeklp@yahoo.com.mx
DOI http://dx.doi.org/10.28960/revmeduas.2007-8013.v12.n4.008
Texto Completo PDFRecibido 22 de septiembre 2022, aceptado 25 de octubre 2022
Abstract
An 18-years-old adolescent with asymmetric mid-ventricular hypertrophic cardiomyopathy, undiagnosed because he did not present symptoms. The electrocardiogram indicated an increase in the QT interval, without arrhythmias; the transthoracic echocardiogram revealed asymmetric mid-ventricular hypertrophy, with mild obstruction of the left ventricular outflow tract; the mitral valve was thickened and calcified. Symptoms were not found because the outflow tract obstruction was not noticeable; as hypertrophy increased, the symptoms appeared. The thickening and calcification of the mitral valve, uncommon for the patient's age, could cause mitral regurgitation or heart failure.
Key Words: Genetic hypertrophic cardiomyopathy, mild outflow tract obstruction, calcification and thickening of the mitral valve.
Resumen
Adolescente de 18 años con miocardiopatía hipertrófica ventricular medio asimétrica, no diagnosticada por no presentar sintomatología. El electrocardiograma indicó aumento del intervalo QT, sin arritmias; el ecocardiograma transtorácico reveló hipertrofia media ventricular asimétrica, con leve obstrucción del tracto de salida del ventrículo izquierdo; la válvula mitral estaba engrosada y calcificada. No se observaron síntomas por no notarse la obstrucción del tracto de salida; a medida que aumentaba la hipertrofia, aparecían los síntomas. El engrosamiento y calcificación de la válvula mitral, infrecuente para la edad del paciente, podría provocar regurgitación mitral o insuficiencia cardíaca.
Palabras Clave: Miocardiopatía hipertrófica genética, Obstrucción leve del tracto de salida, Calcificación y engrosamiento de válvula mitral.
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is an autosomal-dominant disease that is detected in one in every 500 patients; patients with a positive genetic test are often younger than 45 years of age. HCM manifests as left ventricle hypertrophy in the absence of diseases of cardiac or systemic origin. When HCM is asymmetric, it can cause left ventricular outflow tract obstruction, which, in turn, causes valve regurgitation. The diagnosis of HCM is made by echocardiography and cardiac magnetic resonance imaging, which determine the morphological variables of HCM1.
We present a case of a patient with HCM that´s cannot associated with a genetic disease like Noonan’s syndrome, Friedrich's ataxia, Danon or Fabry disease.
CASE PRESENTATION
An 18-years-old male patient with mid-ventricular HCM, with a frequency of symptom occurrence of 5%. The patient was examined in a hospital in the United States in 2017. He presented typical symptoms, including adynamia, chest pain and palpitations, for which symptomatic treatment was prescribed, although the patient did not adhere to this treatment, and no further studies were carried out. The patient denied chronic degenerative diseases. It is unknown whether the patient’s relatives had HCM and therefore whether the HCM was due to genetic causes.
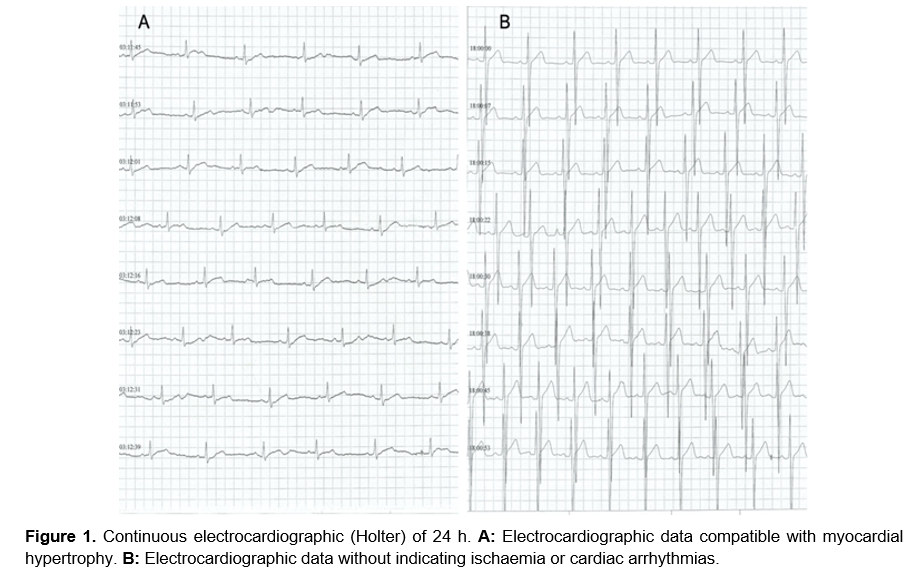
Twelve-lead electrocardiography and transthoracic echocardiography were performed. The electrocardiogram showed increases in the QT interval, without arrhythmias. As the patient did not show chest pain, the band stress test was not performed. Since symptoms and arrhythmias such as ventricular tachycardia can develop suddenly, continuous electrocardiographic monitoring was performed for 24 h (Holter-24 h) to rule them out; however, a normal rhythm and no arrhythmias were observed (Figure 1a), despite the patient exerting effort (walking and light jogging for approximately 30 min). Data compatible with ischaemia were not observed, but data compatible with hypertrophy were observed (Figure 1b). The transthoracic echocardiogram showed asymmetric hypertrophy of the septum due to mid-ventricular obstruction in the apical four-chamber window.
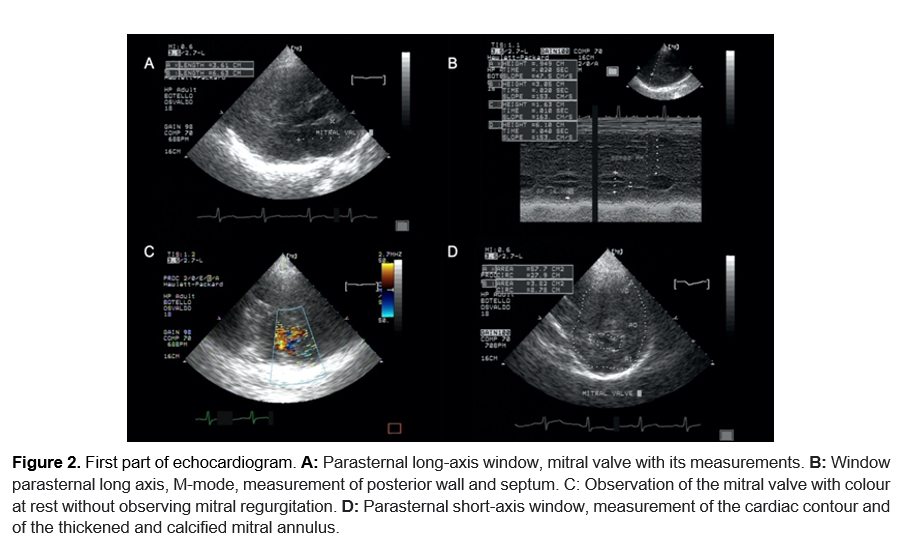
In the long-axis parasternal window, without M mode, the mitral valve was first thickened, subsequently, it was measured to be 36 x 66 mm, thickened and calcified (Figure 2a). In the same window, with M mode, the septum measured 60 mm, and the posterior wall measured 16 mm (Figure 2b). Due to the characteristics of the mitral valve observed at rest, the presence of mitral regurgitation was not identified (Figure 2c). In the short-axis parasternal window, the cardiac circumference was calculated as area (A) = 57.7 cm2 x circumference (C) = 27.9 cm, and the mitral valve opening was calculated as A = 3.82 cm2 x C = 8.78 cm (Figure 2d).
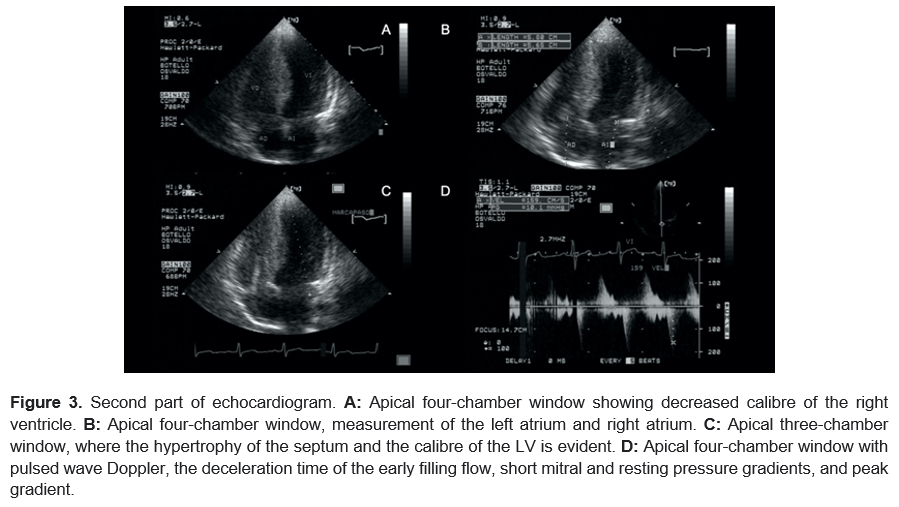
The obstruction reduced the cavity of the right ventricle (Figure 3a). In the apical window, the left atrium measured 58 mm, and the right atrium measured 56 mm (Figure 3b). In the apical four-chamber window, the thickness of the septum and the difference in size between the left and right ventricles were evident (Figure 3c). Finally, pulsed wave Doppler indicated that short mitral early filling flow deceleration time = 159 cm/s. The pressure gradient at rest (peak resting gradient) = 10.1 mm Hg (Figure 3d). The patient's HCM was asymmetric and mid-ventricular, with decreased right ventricular calibre and mild obstruction of the left ventricular outflow tract (<30 mm Hg) due to the size of the septum and the peak gradient at rest2.
DISCUSSION
The adolescent patient presented asymptomatic mid-ventricular hypertrophic cardiomyopathy with a thickening and calcification of the mitral valve and mild obstruction without mitral regurgitation at rest. Mitral valve function was not assessed during exercise. Although the patient reported occasional chest pain when performing activity, during monitoring, no data indicating arrhythmia or ischaemia were recorded. The patient is likely to require a myomectomy in the future due to the degree of hypertrophy. If the peak resting or provoking gradient increases and/or more mitral alterations are found, valve replacement should be considered. In the patient, mid-ventricular HCM was identified, without symptoms, with a thickening and calcification of the mitral valve and mild obstruction without mitral regurgitation at rest. Mitral valve function was not assessed during exercise. Although the patient reported occasional chest pain when performing activity, during monitoring, no data indicating arrhythmia or ischaemia were detected.
In 4460 paediatric HCM patients, patients with genetic causes were younger (8 years) than those without genetic causes (12 years, p <0.0001) (Noonan’s syndrome, Friedrich's ataxia, glycogen storage diseases such as Danon disease, disorders of fatty acid oxidation and lysosomal storage diseases such as Fabry disease). Although the smallest proportion of patients was Hispanic, this ethnic group exhibited the highest proportion of patients with genetic disorders (79%) associated with HCM3.
Among 398 paediatric patients (mean age=14 years), 133 presented symptoms compatible with HCM and arrhythmias, of whom 71 had dyspnoea, 77 had angina and 19 had syncope. Of the 398 paediatric patients, 109 were taking β-blockers for the symptoms described. Among all paediatric patients, the genetic test was positive in 91 of 146 for genes associated with HCM. Only 23% had extreme LV hypertrophy (z score> 6), and 8% had a left ventricular outflow tract> 30 mm; 23 patients had an automatic, and 3 had an appropriate defibrillator shock4. Our patient did not have an automatic defibrillator, as he did not present arrhythmias in the specific risk and benefit assessment. The patient may require myomectomy based on the degree of hypertrophy development, the presence of dyspnoea or chest pain that limit his activity, and mitral regurgitation.
Among the predictors for sudden death caused by severe ventricular arrhythmias in HCM, massive left ventricular hypertrophy (absolute wall thickness ≥ 30 mm) was the most important and, in turn, could be associated with other predictors. In our patient, these predictors could not be used since the LV wall thickness was <30 mm, the peak gradient at rest was 10.1 mm Hg; the obstruction was mild, and no arrhythmias were observed during the evaluation5.
Treatment depends on the degree of hypertrophy and symptoms. The first choice is β-blockers (effective in 60-80% of patients) or calcium channel blockers in patients who do not tolerate β-blockers. Patients with a resting pressure gradient of 30 mm Hg or a provocative gradient >50 mm Hg (severe obstruction) will require interventions such as dual-chamber pacing (10-40% efficacy), septal myomectomy (> 90% efficacy), and septal ablation (70-80% efficiency). Pharmacological treatment should be considered first, and if there is no response or hypertrophy increases, other therapeutic options should be considered6,7. The patient was monitored by continuous electrocardiography to identify or rule out cardiac arrhythmias for 24 h, although he had to be monitored for at least 48 h. Echocardiography should be used to monitor cardiomyopathy and its changes.
REFERENCIAS