Rev Med UAS
Vol. 12: No. 3. Julio-Septiembre 2022
ISSN 2007-8013
Juan David Ceballos-Rojas1, Carlos Rafael Ochoa-Morales1, Francisco Fernando Morales-Sánchez2, Juan Luis Rochin-Terán2, Abraham Verdugo-Rosas2, Fred Valentín Morgan-Ruiz3, Juan Pablo Meza-Espinoza4, Verónica Judith Picos-Cárdenas5, José Alfredo Contreras-Gutiérrez3, Saúl Armando Beltrán-Ontiveros3*
* Correspondencia: Dr. Saúl Armando Beltrán-Ontiveros
Centro de Investigación y Docencia en Ciencias de la Salud. UAS
Dirección: Eustaquio Buelna 91, Col. Burócrata, Código Postal: 80030 Culiacán Rosales, Sinaloa. México.
Teléfono: 667 3068465 saul.beltran@uas.edu.mx
DOI http://dx.doi.org/10.28960/revmeduas.2007-8013.v12.n3.008
Texto Completo PDFRecibido 25 de marzo 2022, aceptado 13 de junio 2022
RESUMEN
La hiperplasia adrenal congénita (HAC) es un trastorno genético autosómico recesivo, causado por la deficiencia de cualquier enzima de la biosíntesis del cortisol, principalmente la 21-hidroxilasa. Hay dos tipos de HAC: clásica y no clásica. La primera es más grave y puede provocar el desarrollo de genitales ambiguos en las niñas, lo que se conoce como pseudohermafroditismo femenino. Debido a la repercusión médica y psicosocial de este trastorno, es de vital importancia su detección temprana, ya que de ello depende establecer un tratamiento oportuno y eficaz. En este sentido, es de gran apoyo el área de la imagenología diagnóstica, ya que estos estudios son altamente sensibles y específicos para el diagnóstico de este trastorno. Aquí, presentamos un caso de pseudohermafroditismo femenino causado por HAC en el que se detectaron hallazgos típicos mediante estudios de imagen.
Palabras clave: Hiperplasia adrenal congénita seudohermafroditismo femenino, genitales ambiguos, resonancia magnética.
Abstract
Congenital adrenal hyperplasia (CAH) is a genetic autosomal recessive disorder caused by deficiency of any enzyme of the cortisol biosynthesis, mainly 21-hydroxylase. There are two types of CAH: classical and non-classical. The former is more serious and can lead to the development of ambiguous genitalia in girls, known as female pseudohermaphroditism. Due to the medical and psychosocial repercussions of this disorder, its early detection is essential, as this is the basis for timely treatment. In this regard, the diagnostic imaging is of great support, because imaging studies are highly sensitive and specific for diagnosis of this disorder. Here, we present a case of female pseudohermaphroditism caused by CAH, in which typical findings were detected by imaging studies.
Key words: Congenital adrenal hyperplasia Female pseudohermaphroditism, ambiguous genitalia, magnetic resonance imaging.
INTRODUCCIÓN
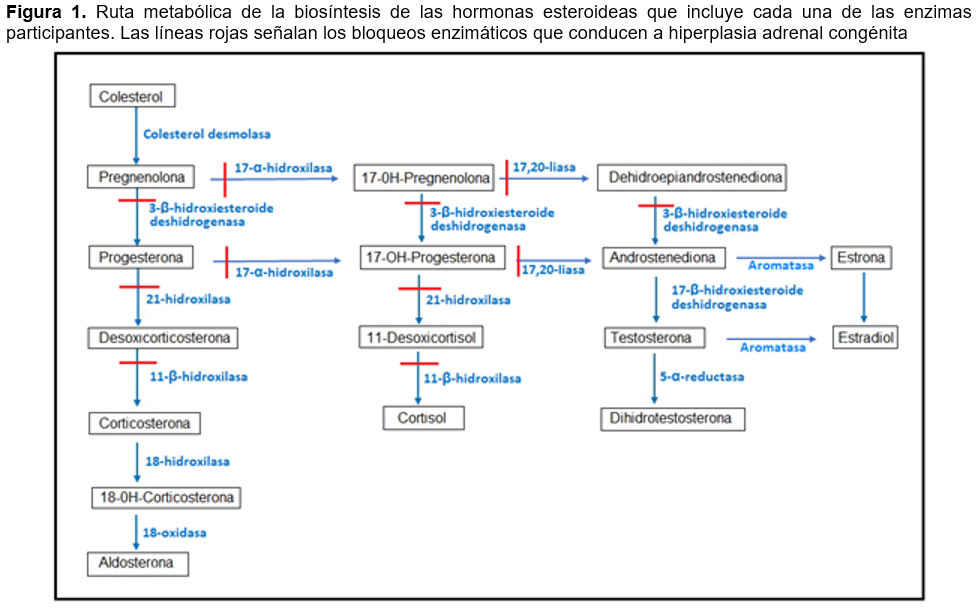
La hiperplasia adrenal congénita (HAC), anteriormente llamada hiperplasia suprarrenal congénita, es un trastorno genético que se produce por alteraciones en la biosíntesis del cortisol y de la aldosterona.1 Se hereda con un patrón de herencia autosómico recesivo y se produce cuando hay deficiencia de una de las enzimas que participan en la biosíntesis de las hormonas esteroideas adrenales (Figura 1).1,2,3
Aproximadamente, el 95% de los casos de HAC se debe a la disminución o ausencia de la enzima 21-hidroxilasa, como consecuencia de mutaciones en el gen CYP21A2.1 Existen diversas mutaciones en este gen, lo que da lugar a variantes enzimáticas con distintos grados de actividad, lo que, a su vez, provoca que la enfermedad presente amplia heterogeneidad clínica.4 Hay dos tipos de HAC: clásica, que se divide en la variante perdedora de sal o en la forma virilizante simple, y no clásica.1,2,5 La HAC clásica es la forma más grave, afecta aproximadamente a 1:15,000.6 La HAC no clásica afecta aproximadamente a 1:100-200 individuos de la población general; sin embargo, los síntomas son más leves, o incluso ausentes.7
Debido a la ausencia de la enzima 21-hidroxilasa, los pacientes no producen cortisol y aldosterona (pacientes con la forma perdedora de sal).1,2,3,5 Los metabolitos precursores, en exceso, se derivan a rutas metabólicas alternas y dan lugar a una producción elevada de andrógenos, lo que puede causar desarrollo anormal de genitales externos en las niñas, provocando fusión de labios mayores, hiperpigmentación y clitoromegalia, simulando los genitales externos masculinos
(pseudohermafroditismo femenino).1,2 También, la HAC puede ser causada por la deficiencia de 17-α-hidroxilasa, 3-β-hidroxiesteroide deshidrogenasa o 11-β-hidroxilasa.2,7
Aunque la incidencia del pseudohermafroditismo femenino es relativamente baja, su repercusión médica y psicosocial es muy importante. Debido a esto es necesario ampliar el conocimiento de sus características clínicas, presentación, diagnóstico y tratamiento, con énfasis especial en los hallazgos de imagen. El papel de la imagenología en el diagnóstico y caracterización es fundamental para un tratamiento médico y quirúrgico oportuno.
Aquí, presentamos un caso de pseudohermafroditismo femenino causado por HAC en el que se detectaron hallazgos típicos a través de estudios imagenológicos.
PRESENTACION DEL CASO
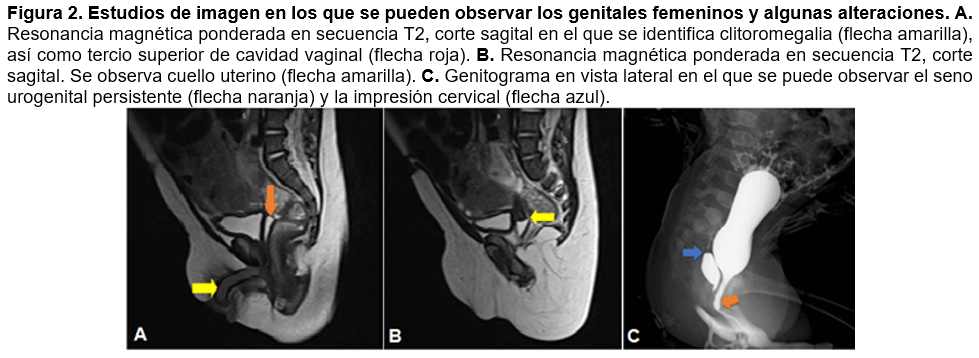
Presentamos el caso de una niña de 6 meses de edad, hija de padres jóvenes no consanguíneos, producto de la primera gestación, a quien, al momento de la valoración de rutina con pediatría, se le detectaron genitales ambiguos, caracterizados por alargamiento del clítoris y fusión de labios mayores, así como ausencia de orificio vaginal. Se le realizó resonancia magnética de pelvis, en la cual se identificó el clítoris, tercio superior de la vagina (Figura 2A) y útero (Figura 2B). Posteriormente, se realizó complemento con genitograma, en el cual se observó seno urogenital persistente (Figura 2C). Además, un análisis de sangre reveló 17-OH-progesterona elevada, por lo que la niña fue diagnosticada con pseudohermafroditismo, secundario a HAC. Sin embargo, los padres no dieron su consentimiento para realizar cariotipo u otros estudios genéticos. Cabe destacar que no se encontraron antecedentes familiares relevantes. Actualmente, la paciente está en tratamiento por el Departamento de Endocrinología Pediátrica y con valoración por el Servicio de Cirugía.
DISCUSIÓN
Como consecuencia del déficit de cortisol, se eleva la concentración de hormona adrenocorticotrópica, lo que estimula la secreción de hormonas adrenales y, a su vez, provoca la HAC.2 Este trastorno produce cambios metabólicos en los individuos que dependen de la forma de presentación y sexo del paciente. En algunas niñas a veces es difícil identificar si los genitales externos son femeninos o masculinos, lo que se conoce como pseudohermafroditismo femenino, el cual también puede ser causado por la exposición de la madre a ciertos fármacos durante el embarazo, como danazol, noretindrona y testosterona.8 Las niñas también pueden presentar vello púbico de aparición temprana, hirsutismo, virilización o irregularidad menstrual en la adolescencia; infertilidad secundaria a anovulación, clítoris aumentado de tamaño en algunos casos e hiperpigmentación de piel y mucosas. Los varones pueden presentar pubertad precoz, caracterizada por crecimiento rápido en la infancia, voz grave, vello púbico y facial. En ambos sexos, la concentración baja de aldosterona puede llevar a sufrir vómito secundario a pérdida de sal, deshidratación y, en casos graves, la muerte.9
Los estudios de imagen, como ultrasonido y resonancia magnética, durante la etapa prenatal son muy importantes para detectar cualquier alteración anatómica durante el desarrollo del feto. Así, el pseudohermafroditismo femenino puede ser detectado a través de estos estudios, ya que mediante ellos se puede observar virilización de los genitales femeninos, como clitoromegalia y fusión de labios mayores; las glándulas adrenales pueden verse normales o aumentadas de tamaño, pero pierden su morfología normal tricorne y adoptan una forma de masa cerebriforme.10,11
En la etapa posnatal, se puede identificar la vagina y caracterizar la longitud del seno urogenital común a través de un genitograma, el cual es confirmado con panendoscopía (cistouretroscopía y vaginoscopía). El ultrasonido permite establecer la presencia de gónadas o derivados de los conductos de Muller; además, es de fácil acceso y no implica radiación. En su protocolo se debe incluir las regiones inguinales, perineales, renales y suprarrenales. La influencia de hormonas maternas en el periodo neonatal hace que el útero y los ovarios sean fáciles de identificar en ultrasonido.12
La fluoroscopía permite identificar la configuración uretral y cualquier comunicación fistulosa con la vagina o el recto. Es frecuente encontrar seno urogenital persistente y el genitograma ayuda a identificar el punto de unión de la uretra y la vagina, así como el nivel del esfínter externo y la impresión cervical, lo cual es muy importante para planear la estrategia quirúrgica. El seno urogenital es el precursor embriológico de la uretra y el tercio distal de la vagina en las mujeres. La malformación de este conduce a la apertura de la uretra y la vagina a través de una sola abertura.12
Para información anatómica detallada se usa la resonancia magnética en secuencias T1 y T2, por su capacidad multiplanar y mejor caracterización tisular. La resonancia magnética es más sensible que el ultrasonido en la evaluación de las gónadas, aunque no es del todo confiable en la evaluación de gónadas intra-abdominales. También es útil para diferenciar el pene del clítoris, ante la ausencia o pobre desarrollo de estructuras de sostén del pene, como el músculo transversal perineal y bulboesponjoso. Además, se puede hacer evaluación renal y adrenal dentro del mismo estudio.12
El tratamiento depende del momento del diagnóstico. En los casos de HAC, diagnosticada prenatalmente, se inicia manejo con dexametasona, idealmente desde la séptima semana de gestación. Por otra parte, cabe mencionar que la HAC se puede diagnosticar desde los dos días de nacimiento, al medir la concentración de 17-OH-progesterona en la sangre. En la etapa posnatal el abordaje farmacológico ideal suele ser con hidrocortisona, pero por su poca disponibilidad comercial se reemplaza por prednisona o dexametasona, ajustada al peso del bebé. Así mismo, se debe complementar el manejo con mineralocorticoides, como fludrocortisona y suplementos de cloruro de sodio, según las necesidades de cada paciente.13 Desde el punto de vista quirúrgico, aún hay controversia sobre cuándo y cómo intervenir. El objetivo de la cirugía es reubicar la vagina en el espacio perineal lo más anatómicamente posible, separando la vagina distal de la uretra y formando un introito adecuado. Si existe la necesidad de reducir el tamaño del clítoris se debe tratar de preservar la función sexual; sin embargo, con el tratamiento hormonal, en la mayoría de los pacientes se produce regresión de la clitoromegalia.14
Por otra parte, independientemente de la gravedad de las manifestaciones clínicas y del abordaje terapéutico que se establezca en los pacientes, es muy importante el asesoramiento genético familiar, puesto que la mayoría de los casos de HAC cumple con un patrón de herencia autosómico recesivo, lo que significa que ambos padres son portadores (obviamente, asintomáticos) de un alelo mutado de un gen que produce enzima que participa en la biosíntesis del cortisol, especialmente CYP21A2, por lo que habrá un riesgo de recurrencia del 25% para la enfermedad, y la pareja debe ser informada del riesgo que se presentará en cada embarazo.
CONCLUSIONES
En este caso de pseudohermafroditismo femenino, ocasionado por HAC, pudimos demostrar la importancia de los estudios de imagenología en el diagnóstico temprano de esta enfermedad.
REFERENCIAS