Rev Med UAS
Vol. 12: No. 3. Julio-Septiembre 2022
ISSN 2007-8013
Juan Pablo Meza-Espinoza1, Juan Ramón González-García2, José Alfredo Contreras-Gutiérrez3, Verónica Judith Picos-Cárdenas4
* Correspondencia: Dra. Verónica Judith Picos-Cárdenas
Laboratorio de Genética, Facultad de Medicina, Universidad Autónoma de Sinaloa, Culiacán, Sin., México.
Tel. 6677538801. Fax. 6677538802. e-mail: veronicapicos@uas.edu.mx
DOI http://dx.doi.org/10.28960/revmeduas.2007-8013.v12.n3.010
Texto Completo PDFRecibido 14 de septiembre 2021, aceptado 13 de mayo 2022
RESUMEN
La leucemia mieloide crónica (LMC) es una enfermedad de la sangre caracterizada por aumento de leucocitos y del tamaño del bazo. Su origen ha sido asociado con exposición a radiación ionizante. En más del 90% de los pacientes se encuentra una translocación cromosómica, la t(9;22)(q34;q11), conocida también como cromosoma Filadelfia, misma que da lugar a la formación del gen híbrido BCR/ABL1, el cual genera una proteína híbrida que tiene la capacidad de transformar a las células afectadas y producir la leucemia. El tratamiento de vanguardia se basa principalmente en fármacos específicos que contrarrestan la actividad de la proteína híbrida de BCR/ABL1, con los que se obtienen excelentes resultados. Sin embargo, es común encontrar pacientes con recaídas, por lo que es fundamental el seguimiento constante de la enfermedad a través de estudios citogenéticos y moleculares.
Palabras clave: LMC, t(9;22)(q34;q11), Ph , transcrito BCR/ABL1, remisión molecular.
Abstract
Chronic myeloid leukemia (CML) is a blood disease characterized by leukocytosis and splenomegaly. Its origin has been associated with exposure to ionizing radiation, and more than 90% of patients are carriers of the t(9;22)(q34;q11) translocation, known also as Philadelphia chromosome, which leads to the formation of the BCR/ABL1 hybrid gene, that has the capacity to transform the affected cells and produce leukemia. Treatment is mainly based on specific drugs against BCR/ABL1 protein, with which excellent results are obtained. However, it is common to find patients with relapses, so constant monitoring of the disease by means of cytogenetic and molecular studies is essential.
Key words:CML, t(9;22)(q34;q11), Ph, BCR/ABL1 transcript, molecular remission
INTRODUCCIÓN
Leucemia mieloide crónica
Definición
La leucemia mieloide crónica (LMC) es un trastorno hematológico mieloproliferativo que se origina a partir de una célula madre transformada de la médula ósea. Se caracteriza por una proliferación excesiva de leucocitos (leucocitosis) con acumulación de células mieloides y sus precursores, lo que ocasiona anemia, aumento anormal de plaquetas (trombocitosis) y del tamaño del bazo (esplenomegalia). Presenta una evolución trifásica: etapa inicial o crónica, relativamente benigna, con duración media de 5 a 7 años, relativamente fácil de controlar, pero si la enfermedad no es tratada de manera oportuna progresa a una fase de inestabilidad, conocida como aceleración y, posteriormente, a una transformación terminal, llamada crisis blástica.1,2
Manifestaciones clínicas
Las complicaciones más comunes de la LMC son fatiga, pérdida de peso, poca tolerancia al esfuerzo, malestar general, anorexia, saciedad temprana, dolor abdominal, esplenomegalia (50-60% de los pacientes), crecimiento del hígado (hepatomegalia, 10-20% de los pacientes), palidez y sudores nocturnos. Entre los hallazgos hematológicos, se detectan cuentas de leucocitos de al menos 25,000/mm2, pero es común encontrar más de 100,000/mm;2 morfológicamente, se pueden observar blastos (células inmaduras), granulocitos en todas las etapas de desarrollo, aumento de basófilos y trombocitosis, leve reticulocitosis, anemia y eritrocitos alterados en tamaño y forma.1,2,3
Etapas de la enfermedad
Fase crónica: Tiene una duración aproximada de 5 a 7 años. Se caracteriza por leucocitosis, fatiga, malestar general, falta de apetito, pérdida de peso, anemia y esplenomegalia.1 Se puede encontrar hasta un 10% de blastos en sangre periférica. Más del 90% de los pacientes que cumplen los criterios clínicos y de laboratorio para el diagnóstico de LMC presenta una anormalidad cromosómica, inicialmente llamada cromosoma Filadelfia (Ph), que corresponde a la translocación t(9;22).2
Fase acelerada: Se le llama así a la etapa de evolución de la enfermedad en la que el paciente muestra una aceleración mieloproliferativa y, como consecuencia, aumenta aún más en sangre periférica la leucocitosis, los basófilos (>20%) y los blastos (10-19%). Los signos y síntomas son más intensos, incluyen trombocitosis (o trombocitopenia: disminución anormal de plaquetas) y aumento de la esplenomegalia.1,2 El 30% de los pacientes presentan otras alteraciones cromosómicas además del cromosoma Ph.4 Esta etapa dura pocos meses.
Crisis blástica: Es propiamente la etapa terminal de la leucemia, por lo cual su duración es de unas semanas, en ésta, se exacerban los signos y síntomas de la enfermedad, se encuentran >20% de blastos en sangre periférica. Se presenta fiebre, hemorragias e infiltración de blastos a tejidos extramedulares, como el SNC.1,2 Esta etapa se caracteriza por gran inestabilidad genómica, por lo que del 60 a 80% de los pacientes presenta múltiples anormalidades cromosómicas.5
Epidemiología
La incidencia anual de LMC varía desde 0.4/100,000 habitantes en algunos países no occidentales hasta 1.75/100,000 en Estados Unidos de América (EUA);6 esta enfermedad predomina ligeramente en varones y representa el 15% de todos los casos nuevos de leucemia en EUA.7 Además, la prevalencia de la LMC se ha estimado en 10-12/100,000 habitantes, con un aumento constante debido a la mejoría en el tratamiento y en la supervivencia de estos pacientes.8 Aunque la enfermedad se presenta en niños y adolescentes, menos del 10% de los casos ocurre en pacientes de 1 a 20 años y representa sólo el 3% de los casos de leucemia en niños.2 La supervivencia global a 5 años es del 85% para pacientes diagnosticados en fase crónica. La supervivencia relativa es de aproximadamente 90% en pacientes de 75 a 80 años de edad. Por lo tanto, en países donde los inhibidores de tirosina cinasa (TKI, del inglés tyrosine kinase inhibitor) están disponibles, la mayoría de los pacientes con LMC tienen una esperanza de vida cercana a la de la población normal.8 En EUA la tasa de mortalidad es menor a 0.3/100,000 habitantes por año.6 En México, la LMC corresponde aproximadamente al 10% de todas las leucemias, la edad promedio al diagnóstico es de 45.8 años y alrededor de 57.8% de afectados son varones;9 esta frecuencia es muy similar a la reportada en EUA, la cual corresponde al 56.5%.10 En México, la tasa de mortalidad por LMC en el año 2004, fue reportada en 3/100,000 habitantes.11
Etiología
No se conoce con exactitud la causa de la LMC; sin embargo, se sabe que gente expuesta a grandes dosis de radiación ionizante tiene mayor riesgo de padecer la enfermedad, tal es el caso de la población japonesa, expuesta a la radiación de las explosiones de las bombas atómicas12 y de pacientes que han sido tratados con radiaciones para diferentes trastornos, como linfoma no Hodgkin,13 cáncer testicular14 y hemorragias del útero.15 También se ha documentado que la exposición al benceno aumenta el riesgo de padecer LMC.16
Fisiopatología
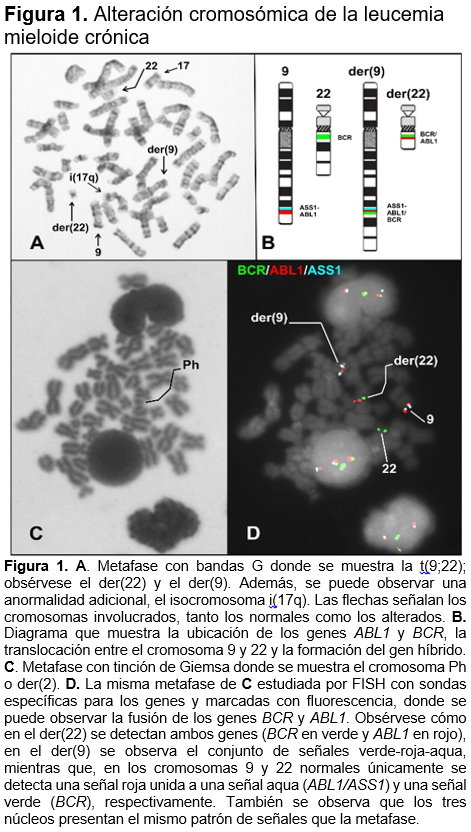
La LMC es originada por una alteración cromosómica conocida como translocación entre el cromosoma 9 y el cromosoma 22, t(9;22)(q34;q11), (Figura 1A), este arreglo cromosómico da como resultado la formación de un gen híbrido, producto de la unión del gen BCR, ubicado en el cromosoma 22, y del gen ABL1, localizado en el cromosoma 9.1,2 El gen ABL1 se expresa en todas las células del cuerpo y produce una enzima de 145 kilodaltones (kDa) con actividad de tirosina cinasa, por lo que cataliza reacciones de fosforilación, lo cual es importante para regular diversas funciones celulares, particularmente, el ciclo celular.17 El gen BCR también se expresa en todas las células y produce una proteína de 160 kDa que tiene actividad de serina/treonina cinasa,1,2 y de esta manera, activa diversas proteínas importantes en la proliferación celular, tales como NF-kB, PDGF, RAS, RAF-1, ERK1/2 y ELK1.18 El gen híbrido BCR/ABL1 se localiza en el cromosoma derivativo 22, también conocido como cromosoma Filadelfia (Ph) (Figura 1B-D). Una vez que este gen híbrido es transcrito, se traduce en la proteína BCR/ABL1, la cual tiene mayor actividad tirosina cinasa que la proteína normal de ABL1, por lo que adquiere la capacidad de autofosforilarse y mantenerse activa de manera permanente, lo que le confiere el potencial de transformar a las células afectadas y activar diversas vías de señalización mitogénica, que originan una proliferación celular descontrolada.18
Gen BCR/ABL1
En general, se han descrito tres regiones de agrupamiento de puntos de ruptura en el gen BCR: mayor (M-bcr), menor (m-bcr) y micro (µ-bcr).18 En la mayoría de los pacientes con LMC, los puntos de ruptura ocurren en la región M-bcr, que abarca aproximadamente 5.8 kilobases (kb) entre los exones e13 y e15 (antes llamados b2-b4).19 La región m-bcr se localiza en un área de 54.4 kb entre los exones e2’ y e2, mientras que la µ-bcr ocurre en el exón e19,18 (Figura 2A). Por su parte, los puntos de ruptura en el gen ABL1 se distribuyen a lo largo de una región de >300 kb del extremo 5’, pudiendo ocurrir antes del primer exón alternativo Ib, entre los exones Ib y Ia, o bien, después del exón Ia, (Figura 2B). Sin embargo, independientemente del punto de ruptura, siempre resultan en la fusión del exón 2 (a2).18,19 Así, los genes y transcritos híbridos dependen de la localización del punto de ruptura el gen BCR. Las rupturas en la región M-bcr generan transcritos e13a2 y e14a2 (anteriormente conocidos como b2a2 y b3a2, respectivamente); el transcrito e14a2 es 75 bases más grande que el transcrito e13a2, pero ambos codifican una proteína de 210 kDa.20 Las rupturas en la región m-bcr generan un transcrito e1a2 que se traduce en una proteína de 190 kDa, la cual es más frecuente en leucemia linfoblástica aguda.21 y las rupturas en la región µ-bcr dan lugar al transcrito e19a2 (Figura 2C), que codifica una proteína de 230 kDa y se encuentra en casos de LMC con maduración neutrofílica y/o trombocitosis.22 Ocasionalmente, se han descrito otros transcritos híbridos, tales como b2a3, b3a3, e1a3, e6a2 y e2a2.18
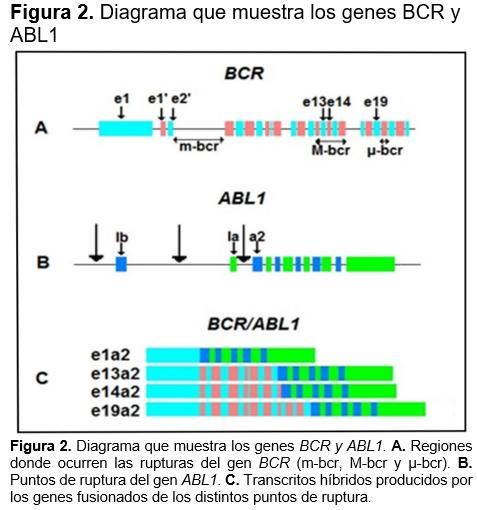
Por lo general, los pacientes con LMC presentan cualquiera de los transcritos e13a2 o e14a2; sin embargo, en el 5% de los casos se encuentran ambos.23 En un estudio realizado en 93 pacientes con LMC del occidente de México, la frecuencia de estos transcritos fue de 48% para e14a2, 40% para e13a2 y en el 12% de los pacientes se detectaron ambos transcritos.24 Cualquiera de los transcritos híbridos produce una enzima con actividad de tirosina cinasa aumentada, que activa una serie de procesos, también conocidos como vías de señalización, que afectan el equilibrio del ciclo celular y dan a las células transformadas ventajas de crecimiento y proliferación, ya que disminuye la adherencia celular, se altera el control de la proliferación celular y se inhibe la muerte normal de las células (con incremento de su tiempo de vida), lo que conduce al proceso neoplásico.18 Resulta de gran de importancia pronóstica la determinación del tipo de transcrito híbrido BCR/ABL1 en cada paciente con LMC, ya que, se ha documentado que los pacientes que expresan el transcrito e13a2 tienen una respuesta molecular más lenta y, a largo plazo, resultados terapéuticos más pobres que los pacientes que expresan el transcrito e14a2. Sin embargo, los pacientes con el transcrito e14a2 tienen más probabilidad de presentar trombocitosis.25
Evolución clonal
La gran mayoría de los pacientes con LMC (más del 90%) presentan la translocación cromosómica t(9;22)(q34;q11), la cual, sin duda, es el marcador citogenético de la enfermedad en fase crónica. Sin embargo, durante la evolución de la enfermedad, ya sea en fase acelerada o crisis blástica, se presentan anormalidades cromosómicas adicionales (ACA), entre las que se encuentran principalmente trisomía del cromosoma 8, un segundo cromosoma Ph, isocromosoma i(17q10), trisomía 19, trisomía 21 y pérdida del cromosoma Y.5 En un estudio en pacientes mexicanos realizado antes del inicio de la terapia, las ACA que se encontraron con mayor frecuencia fueron un tanto diferentes en frecuencia con las reportadas en otras poblaciones; estas fueron tetraploidías, trisomía 8, inversión inv(3)(q21q26), cromosoma Ph adicional e isocromosoma i(17q). Estas diferencias pudieran indicarnos que el patrón de evolución de la enfermedad está relacionado con influencias medioambientales y del componente genético de cada población.26
Las ACA también varían de acuerdo al tratamiento que se administra a los pacientes. En este sentido, cabe mencionar que algunos medicamentos utilizados anteriormente en el tratamiento de la LMC y que en la actualidad están en desuso, se asocian con la aparición de ACA específicas. Así, la trisomía 8 es más frecuente en pacientes tratados con busulfán que en quienes recibieron hidroxiurea; mientras que las ACA que se observan después de tratamiento con interferón-α (IFN-α) o trasplante de médula ósea (TMO) no son comunes, parecen aleatorias y ocasionalmente transitorias.5 Las ACA tienen implicaciones pronósticas importantes, por ejemplo; el i(17q), las anomalías de 3q26.2 y las aberraciones complejas, se han asociado con resultados desfavorables, mientras que la presencia de un cromosoma Ph adicional, la trisomía 8, la pérdida del cromosoma Y, y la deleción intersticial en el der(9), no tienen un impacto significativo en el pronóstico de pacientes bajo terapia con TKIs.27
Diagnóstico citogenético y molecular
Si el paciente reúne los criterios clínicos y hematológicos de LMC, se debe realizar un cariotipo en médula ósea para ratificar el diagnóstico, el cual será confirmatorio si se encuentra la translocación t(9;22)(q34;q11) (Figura 1A). En caso de no detectar esta anormalidad o que la cantidad y calidad de las células metafásicas no sean apropiadas para obtener un análisis confiable del cariotipo, se recomienda un estudio de hibridación in situ fluorescente (FISH, del inglés Fluorescence in situ hibridization), el cual se basa en la unión de sondas específicas de los genes BCR y ABL1 marcadas con una molécula fluorescente (Figura 1D) y es útil para diagnosticar la fusión BCR/ABL1 en pacientes con diagnóstico clínico de LMC y negativos para la t(9;22).28 Otra técnica que se utiliza para el diagnóstico de la LMC es la RT-PCR (Reacción en cadena de la polimerasa con transcripción reversa, del inglés Reverse transcription polymerase chain reaction), la cual es una técnica muy sensible y específica, que se basa en la amplificación de ADN complementario (obtenido a partir del ARN mensajero), con la que se puede analizar la presencia de los transcritos híbridos BCR/ABL1 en muestras de sangre.29 El método de RT-PCR puede ser cualitativo o cuantitativo (este último: qRT-PCR) el primero sólo proporciona información sobre la presencia de transcritos, por lo que puede ser de utilidad para el diagnóstico inicial; el segundo evalúa la cantidad de los transcritos y se usa en pacientes en remisión, ya que es ideal para la detección de enfermedad residual.30
Tratamiento
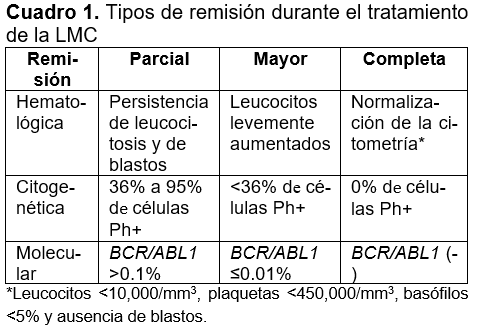
La meta del tratamiento siempre es lograr que el paciente obtenga no sólo la mejoría clínica, además de la remisión hematológica, sino que es vital la remisión citogenética y molecular (Cuadro 1), lo que aumenta la posibilidad de que se logre un estado de enfermedad libre de tratamiento, para lo cual es muy importante vigilar la remisión molecular del paciente a mediano y largo plazo.31,32 En la actualidad, se recomienda que el tratamiento de primera línea sea con un TKI; no obstante, en pacientes sintomáticos en fase crónica con recuentos elevados de leucocitos o plaquetas, puede administrarse hidroxiurea mientras se confirma citogenética o molecularmente el diagnóstico de LMC.31 A continuación, se muestran recomendaciones para el diagnóstico y seguimiento de pacientes con LMC.31,32
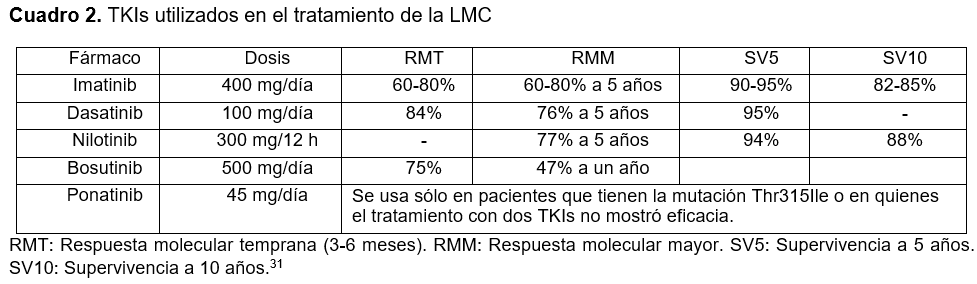
Antes de la introducción de los TKIs, el IFNα era el tratamiento de elección para el control de esta enfermedad, y el TMO era la única opción de curación. Sin embargo, el mayor obstáculo que enfrenta el TMO es la enfermedad injerto contra huésped, que consiste en una complicación multisistémica potencialmente mortal que puede ocurrir después de un TMO. Esta complicación se puede presentar en dos formas: aguda y crónica; la primera, aparece de dos a seis semanas después del trasplante y afecta aproximadamente al 39% de los pacientes que reciben trasplantes de donadores completamente histocompatibles (HLA idéntico) y hasta en 59% de los trasplantes de donadores no emparentados;33 la segunda, se presenta del 30% al 80% de los pacientes que sobreviven seis meses o más después del trasplante y es la principal causa de muertes que ocurren después de dos años del trasplante, aún sin haber recaída.34 El factor principal que influye en la efectividad de un TMO es la fase de la enfermedad al momento de la intervención, así como si se realiza dentro del primer año, lo que mejora las probabilidades de una mayor supervivencia.35 Además, los pacientes más jóvenes tienen mejores resultados. Generalmente, los pacientes en fases avanzadas (acelerada y blástica) no responden bien; aun así, el TMO sigue siendo el tratamiento de elección para pacientes en fases avanzadas de la enfermedad.36 Los TKIs, cuyo blanco terapéutico es la proteína BCR/ABL1, han mostrado buena eficacia en el tratamiento de la LMC, ya que se logran altas tasas de remisión citogenética, y la supervivencia a ocho años es del 87%.37 Entre los TKIs utilizados se encuentran el imatinib, dasatinib, nilotinib, bosutinib y ponatinib, con los cuales ha tenido buena respuesta molecular y supervivencia a largo plazo31,38 (Cuadro 2). Generalmente, el imatinib es el fármaco de primera elección, pero si el paciente muestra resistencia (no logra una respuesta hematológica y citogenética adecuada) o intolerancia, entonces se administra dasatinib, nilotinib, bosutinib o ponatinib, o se valoran otras estrategias de terapia, como la administración de IFN-α en combinación con algún TKI39 o el TMO.
Mutaciones en los dominios de la proteína BCR/ABL1
Actualmente, para el tratamiento de primera línea se utiliza un TKI como el imatinib, el cual es el más utilizado. Sin embargo, en algunas ocasiones se desarrolla una resistencia al tratamiento, manifestada como una pobre respuesta hematológica y/o citogenética, y un regreso a las principales características clínicas de la enfermedad. Se ha observado que dicha resistencia está relacionada con la adquisición de mutaciones en el gen híbrido BCR/ABL1, las cuales modifican el sitio de unión del TKI a la proteína hibrida e impiden el efecto inhibitorio del fármaco sobre la proteína. De lo anterior, se desprende que un análisis de mutaciones del gen BCR/ABL1 es necesario cuando hay resistencia al tratamiento de TKI. Tales análisis también proporcionan información muy importante para la elección del TKI ideal para contrarrestar la resistencia. En pacientes con la mutación Thr315Ile, resistentes a la mayoría de los TKIs, el único tratamiento que ha mostrado mejoría es el ponatinib.40 Otras mutaciones, aunque no confieren una resistencia completa, influyen en la intensidad de la respuesta. Por ejemplo, Glu255Lys/Val es relativamente resistente a nilotinib, mientras que Phe317Lys/Ile es resistente a dasatinib. Asimismo, es rara la RCC en pacientes con la mutación Glu255Val/Lys tratados con nilotinib y en pacientes con la mutación Phe317Lys/Ile tratados con dasatinib. El diagnóstico de resistencia a imatinib tiene importantes implicaciones pronósticas y no debe tomarse a la ligera. La resistencia a cualquier medicamento obliga a reevaluar las opciones terapéuticas de TKIs disponibles31,32 (Cuadro 3).

Vigilancia de la enfermedad
La citometría hemática debe realizarse semanalmente hasta que se estabilice el número de células. Una vez que se alcanza la remisión hematológica completa (RHC), el seguimiento continúa con el cariotipo de médula ósea, FISH y/o estudios de qRT-PCR, para valorar la respuesta citogenética y molecular. Una RCC se asocia con mayor supervivencia a largo plazo.41 Además, el cariotipo en médula ósea es importante para analizar la existencia de ACA y detectar de manera oportuna la evolución de la enfermedad. Se recomienda a los 6, 12 y 18 meses, o hasta que se haya alcanzado la RCC. Hay una serie de recomendaciones de metas terapéuticas ampliamente aceptadas por la comunidad científica para determinar si el tratamiento con imatinib está dando resultados. Estas recomendaciones clasifican las respuestas en óptimas, parciales y fallidas32 (Cuadro 4).
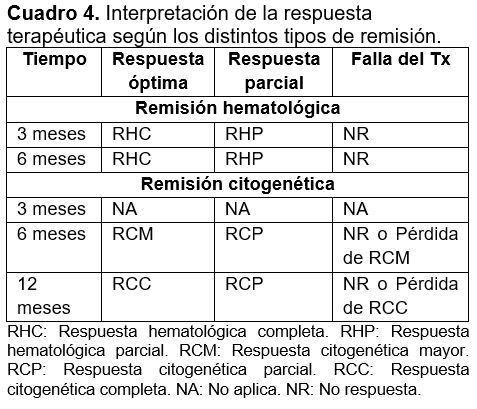
Una falla implica que no se logró la remisión esperada para el tiempo de tratamiento indicado, por lo que se recomienda un cambio de estrategia terapéutica.
Una respuesta óptima implica que el paciente obtiene buenos resultados con imatinib. En caso de una respuesta parcial, se puede continuar con el imatinib, pero es probable que sea necesario realizar ajustes. Una RHC indica que el paciente ha logrado normalizar y mantener estable las células de la sangre. Una respuesta hematológica parcial (RHP) indica que el paciente aún persiste con un número incrementado de células. La respuesta citogenética mayor (RCM) equivale <36% de células Ph+; la respuesta citogenética parcial (RCP) indica que persisten de 36% a 95% de células Ph+. Sin embargo, cabe mencionar que el determinar con exactitud el grado de respuesta citogenética se complica por diversos factores, entre los que destacan el obtener un número suficiente de metafases para el análisis, la calidad de las mismas y el arduo trabajo que implica analizar al menos 100 metafases por cada paciente.
En este sentido, cobra importancia el uso de la FISH, mediante la cual se pueden analizar más de 500 metafases en poco tiempo y así proporcionar más información sobre la respuesta citogenética.
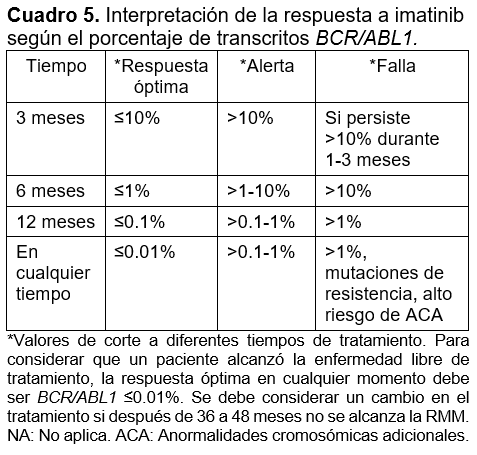
Por otra parte, los estudios de qRT-PCR son muy importantes para analizar la respuesta molecular, la cual se evalúa, según la Escala Internacional, como la relación entre los transcritos de BCR/ABL1 y los transcritos de ABL1 (gen control), y se expresa como porcentaje de BCR/ABL1 en una escala logarítmica [(BCR-ABL1/ABL1) x 100], donde 1%, 0.1%, 0.01%, 0.0032% y 0.001% corresponden a una disminución de 2, 3, 4, 4.5 y 5 logaritmos, respectivamente, por debajo de la línea de base estandarizada de referencia que se utilizó en el International Randomized Study of Interferon and STI571 (IRIS).
Un nivel de transcritos BCR/ABL1 ≤1% equivale a una RCC. Un nivel de BCR/ABL1 ≤0.1% se define como RMM o MR3. Un nivel de BCR/ABL1 ≤0.01% o una enfermedad indetectable en el ADN complementario con >10,000 transcritos de ABL1 se define como MR4. Un nivel de transcritos BCR/ABL1 ≤0.0032% o enfermedad indetectable en el ADN complementario con >32,000 transcritos de ABL1 se define como MR4.5.
No obstante, es preferible el uso del término “leucemia molecularmente indetectable” que el de “respuesta molecular completa”.31
El cuadro 5 muestra respuestas a imatinib según el porcentaje de BCR/ABL1.
Actualidad y perspectivas de la LMC en nuestro país
En general, en nuestro país, la incidencia de LMC no está completamente documentada, ya que no existe un programa nacional que registre la ocurrencia de esta enfermedad,42 y, aunado a que algunos pacientes no son diagnosticados, se desconoce realmente la prevalencia de personas afectadas. Por otra parte, por ser México un país en vías de desarrollo, el sistema nacional de salud carece de la infraestructura y equipamiento necesario para realizar el diagnóstico, tratamiento y seguimiento de los pacientes con LMC en la mayor parte del territorio nacional. Si acaso, las principales ciudades del país son las que cuentan con laboratorios institucionales especializados para hacer el diagnóstico citogenético y molecular en estos pacientes, pero sin que existan recursos económicos ni de infraestructura suficientes, tampoco personal capacitado para realizar de manera sistemática este tipo de análisis. Y, aunque existen laboratorios privados, el precio de sus servicios es elevado y muy difícil de ser cubierto por la mayoría de los pacientes. Además, los medicamentos son costosos y deben ser utilizados sistemáticamente a largo plazo, por lo que no todos los pacientes tienen acceso al tratamiento, y menos aquéllos que no están adscritos a alguna institución de salud pública. Y qué decir acerca de los tratamientos de última generación, los cuales son utilizados en los países del primer mundo; en México sigue siendo el imatinib el único TKI disponible para el tratamiento de la LMC, independientemente del estadio de la enfermedad, o de si se observa resistencia o intolerancia al fármaco. Incluso, el análisis de mutaciones de la proteína de BCR/ABL1, indispensable para la elección del esquema de tratamiento en pacientes con resistencia a TKIs de primera o segunda línea, ha sido pobre, y sólo escasos estudios en un programa de “uso compasivo” han sido realizados.42 Por todo lo anterior, es evidente que se requieren cambios importantes sobre el abordaje de la LMC en nuestro país, ya que sólo así se logrará una mayor proporción de pacientes con enfermedad libre de tratamiento y supervivencia a largo plazo.
Conclusiones
No hay duda que desde la introducción de los TKI para el tratamiento de la LMC, en el año 2001, se incrementó la supervivencia de los pacientes. Antes de ese tiempo, esta enfermedad era prácticamente incurable y los pacientes tenían, en general, una esperanza de vida corta; la tasa de supervivencia a 10 años era sólo del 20%; el imatinib aumentó esta tasa hasta 80%-90%.6,43 En la actualidad, se considera que la mayoría de los pacientes con LMC pueden llevar una vida prácticamente normal si se les administra esta terapia. Así, el objetivo del tratamiento ha pasado de prolongar la supervivencia a mantener la calidad de vida de los pacientes.1 Sin embargo, aunque con la terapia de los TKIs se logran RCC y RMM, en muchos de esos casos persisten células leucémicas residuales, por lo que es de suma importancia el seguimiento de ellos para evitar recaídas. Aquí, los estudios citogenéticos y los análisis de qRT-PCR son de suma importancia para este fin. Por otra parte, no debemos olvidar que del 10 al 15% de los individuos tratados llegan a ser resistentes a los TKIs disponibles,31 por consiguiente, sigue siendo un reto lograr que todos los pacientes alcancen una enfermedad libre de tratamiento a largo plazo.
REFERENCIAS